Aktive ingredienser: Glutation
TAD® 600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning
Hvorfor brukes Tad 600? Hva er den til?
FARMAKOTERAPEUTISK KATEGORI
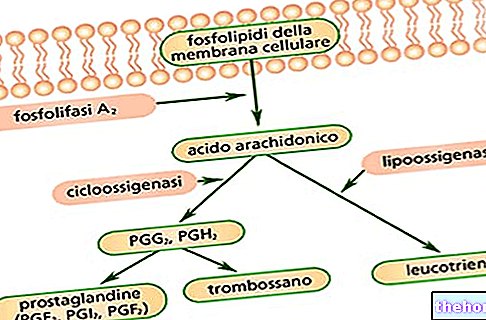
Glutation er et fysiologisk tripeptid som griper inn i mange biologiske prosesser og spiller en viktig rolle i avgiftningsreaksjoner, som beskytter celler mot skadelig virkning av xenobiotiske midler, miljø- og intracellulære oksidanter og stråling.
Parenteralt administrert glutation tilhører den farmakoterapeutiske gruppen av motgift.
TERAPEUTISKE INDIKASJONER
Profylakse av nevropati etter cellegiftbehandling med cisplatin eller analoger.
Kontraindikasjoner Når Tad 600 ikke skal brukes
Overfølsomhet overfor virkestoffet.
Forholdsregler for bruk Hva du må vite før du bruker Tad 600
Ingen spesielt.
Interaksjoner Hvilke medisiner eller matvarer kan endre effekten av Tad 600
Ingen tilfeller av legemiddelinteraksjoner med glutation er beskrevet. Ved anbefalte doser forstyrrer ikke TAD den terapeutiske aktiviteten til det kjemoterapeutiske middelet.
Advarsler Det er viktig å vite at:
De tilgjengelige dataene indikerer at glutation, på grunn av dets natur som et stoff som er fysiologisk tilstede i celler, ikke gir noen bivirkninger hos gravide eller ammende. Dyrestudier indikerer ikke direkte eller indirekte skadelige effekter i forhold til graviditet, embryo-foster utvikling, fødsel eller postnatal utvikling.
Sikkerhet og effekt hos barn er ikke fastslått.
Advarsel: Produktene for parenteral bruk må inspiseres visuelt før administrering, når beholderen eller løsningen tillater det, for å oppdage mulig forekomst av partikler eller avvikende fargestoffer. Skal ikke brukes hvis det er uklarhet eller bunnfall.
FORHOLD UTILGJENGELIG FOR BARN.
Dosering og bruksmåte Hvordan bruke Tad 600: Dosering
Dosering
Den generelt anbefalte daglige dosen av TAD hos pasienter som får cisplatin eller analog kjemoterapi er 1,5 g / m2 (tilsvarende 2500 mg) administrert sakte intravenøst. Dosen er imidlertid avhengig av pasientens alder, vekt og kliniske tilstand, og bør også være korrelert med dosen og doseringsregimet for kjemoterapien Ved administrering av glutation i kombinasjon med cellegift, bør intravenøs infusjon av TAD forekomme innen 15 til 30 minutter før starten av cellegift.
Ved langvarige behandlinger kan de laveste dosene av produktet (600 mg) brukes til å administreres intramuskulært eller sakte intravenøst.
Administrasjonsmåte
Rekonstituer oppløsningen i hetteglasset med pulver ved å trekke ut vannet i hetteglasset med løsningsmiddel ved hjelp av en sprøyte utstyrt med en passende nål. Fjern aluminiumsfliken på hetteglasset og desinfiser proppen med en bomullspinne fuktet i alkohol, og sett deretter sprøytenålen inn inn i hetteglasset gjennom midten av gummiproppen og led vannstrømmen til glassveggen i hetteglasset. Rist forsiktig for å lette fullstendig oppløsning, og administrer deretter oppløsningen som er oppnådd langsomt intramuskulært eller intravenøst.
Viktig: Bruk umiddelbart etter at beholderne er åpnet. Den rekonstituerte oppløsningen må være klar og fri for synlige partikler. Den er for en uavbrutt administrering og eventuelle rester kan ikke brukes.
Overdosering Hva du skal gjøre hvis du har tatt for mye Tad 600
Det er ikke rapportert tilfeller av overdosering. Om nødvendig kan symptomatiske behandlinger brukes.
Bivirkninger Hva er bivirkningene av Tad 600
Utslett har blitt rapportert svært sjelden etter intramuskulær administrering og forsvant ved seponering av behandlingen. Det er også rapportert smerter på injeksjonsstedet.
Som med alle parenterale løsninger kan feberreaksjoner, infeksjoner på injeksjonsstedet, venøs trombose eller flebitt forekomme ekstravasal diffusjon.
I tilfelle en umiddelbar bivirkning under intravenøs infusjon, avbryt administrasjonen og, hvis mulig, behold den resterende væsken som ikke administreres for mulige tester.
Overholdelse av instruksjonene i dette pakningsvedlegget reduserer risikoen for uønskede effekter.
Det er viktig å informere lege eller apotek om enhver uønsket effekt, selv om det ikke er beskrevet i pakningsvedlegget.
Utløp og oppbevaring
Se utløpsdatoen som er trykt på pakken. Denne datoen refererer til produktet i intakt emballasje, riktig lagret.
Advarsel: ikke bruk medisinen etter utløpsdatoen som er angitt på pakningen.
Sammensetning og farmasøytisk form
SAMMENSETNING
Hvert hetteglass med pulver inneholder:
Aktiv ingrediens: glutation natriumsalt 646 mg, lik glutation 600 mg. Hvert hetteglass med væske inneholder:
Hjelpestoff: vann til injeksjonsvæsker.
Den rekonstituerte oppløsningen inneholder 150 mg / ml glutation.
LEGEMIDDELFORM OG INNHOLD
Pulver og væske til injeksjonsvæske, oppløsning.
Kartongen inneholder 10 hetteglass med 600 mg pulver og 10 hetteglass med oppløsningsmiddel på 4 ml.
Kildepakningsvedlegg: AIFA (Italian Medicines Agency). Innhold publisert i januar 2016. Informasjonen som er tilstede er kanskje ikke oppdatert.
For å få tilgang til den mest oppdaterte versjonen, er det lurt å gå til nettstedet til AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttig informasjon.
01.0 LEGEMIDLETS NAVN
TAD
02.0 KVALITATIV OG KVANTITATIV SAMMENSETNING
600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning
Ett hetteglass med pulver inneholder:
Aktiv ingrediens: glutation natriumsalt 646 mg lik glutation 600 mg.
Den rekonstituerte oppløsningen inneholder 150 mg / ml glutation.
2500 mg / 25 ml pulver og væske til infusjonsvæske, oppløsning
En flaske pulver inneholder:
Aktiv ingrediens: glutation natriumsalt 2680 mg lik glutation 2500 mg.
Den rekonstituerte oppløsningen inneholder 100 mg / ml glutation.
For hjelpestoffer, se pkt.6.1.
03.0 LEGEMIDDELFORM
Pulver og væske til injeksjonsvæske, oppløsning.
Pulver og væske til infusjonsvæske, oppløsning.
04.0 KLINISK INFORMASJON
04.1 Terapeutiske indikasjoner
Profylakse av nevropati etter cellegiftbehandling med cisplatin eller analoger.
04.2 Dosering og administrasjonsmåte
Den generelt anbefalte daglige dosen av TAD hos pasienter som får cisplatin eller analog kjemoterapi er 1,5 g / m2 (tilsvarende 2,5 g) administrert sakte intravenøst. Dosen er imidlertid avhengig av pasientens alder, vekt og kliniske tilstand, og bør også korreleres med dosen og doseringsregimet for cellegift.
Ved administrering av glutation i kombinasjon med cellegift, bør intravenøs infusjon av TAD skje innen 15 - 30 minutter før starten av cellegift.
Ved langvarige behandlinger kan de laveste dosene av produktet (600 mg) brukes til å administreres intramuskulært eller sakte intravenøst.
Se avsnitt 6.6 for fremstillings- og administrasjonsmetoder.
04.3 Kontraindikasjoner
Overfølsomhet overfor virkestoffet.
04.4 Spesielle advarsler og passende forholdsregler for bruk
Sikkerhet og effekt hos barn er ikke fastslått.
Merk følgende: Produkter for parenteral bruk må inspiseres visuelt før administrering, når beholderen eller løsningen tillater det, for å oppdage tilstedeværelse av partikler eller unormal farge. Skal ikke brukes hvis det er uklarhet eller bunnfall.
FORHOLD UTILGJENGELIG FOR BARN
04.5 Interaksjoner med andre legemidler og andre former for interaksjon
Tilfeller av legemiddelinteraksjoner med glutation er ikke beskrevet i litteraturen.
Ved anbefalte doser forstyrrer ikke TAD den terapeutiske aktiviteten til det kjemoterapeutiske middelet.
04.6 Graviditet og amming
Tilgjengelige data indikerer at glutation, på grunn av dets natur som et stoff som er fysiologisk tilstede i celler, ikke gir opphav til uønskede effekter hos gravide eller ammende kvinner Prekliniske studier indikerer ikke direkte eller indirekte skadelige effekter på graviditet., Embryofoetal utvikling, fødsel eller postnatal utvikling (se pkt. 5.3).
04.7 Påvirkning av evnen til å kjøre bil og bruke maskiner
TAD har ingen eller ubetydelig påvirkning på evnen til å kjøre bil eller bruke maskiner.
04.8 Bivirkninger
Utslett har blitt rapportert svært sjelden etter intramuskulær administrering og forsvant ved seponering av behandlingen. Det er også rapportert smerter på injeksjonsstedet.
Som med alle parenterale løsninger kan feberreaksjoner, infeksjoner på injeksjonsstedet, venøs trombose eller flebitt forekomme ekstravasal diffusjon.
I tilfelle en umiddelbar bivirkning under intravenøs infusjon, avbryt administrasjonen og, hvis mulig, behold den resterende væsken som ikke administreres for mulige tester.
04.9 Overdosering
Det er ikke rapportert tilfeller av overdosering. Om nødvendig kan symptomatiske behandlinger brukes.
05.0 FARMAKOLOGISKE EGENSKAPER
05.1 Farmakodynamiske egenskaper
ATC: V03AB32 - Motgift.
Glutation (GSH) er et fysiologisk tripeptid sammensatt av glutaminsyre, cystein og glycin, som griper inn i mange biologiske prosesser og spiller en viktig rolle i avgiftningsreaksjoner, og beskytter celler mot skadelig virkning av xenobiotiske midler, miljøoksidanter og intracellulære (gratis radikaler, reaktive oksygenmellomprodukter) og stråling. Prekliniske og kliniske studier har vist glutationons beskyttende rolle i mange patologiske situasjoner som forårsaker celleskader, for eksempel forgiftning fra stoffer som etylalkohol, paracetamol, salisylsyre, fenobarbital, trisykliske antidepressiva, organo-fosforsyre insektmidler, etc. Det har også blitt observert at mange kjemoterapeutiske midler reduserer vev og intracellulære nivåer av endogent GSH, noe som forverrer tilstanden til tumorindusert oksidativt stress.
Spesielt når det gjelder nevrotoksisitet fra kjemoterapeutiske legemidler som cisplatin og derivater, antas det at det skyldes akkumulering av platina i det perifere nervesystemet, spesielt i de bakre nerverotrenganglene. I tilfelle av oksaliplatin ser det ut til at akkumulering av platina skyldes langsommere eliminering i stedet for økt avsetning.Dette antyder bruk av midler som glutation som er i stand til å forhindre den første akkumuleringen av platina i de bakre nerverotrangliene.
Mange kliniske studier har bekreftet denne effekten av glutation: de viser hvordan infusjon av glutation før administrering av antiblastik hos pasienter med eggstokkreft, magekreft og tykktarmskreft gir en effektiv beskyttelse mot nefro og nevrotoksisitet forårsaket av cisplatin og oksaliplatin, slik at, om nødvendig oppnåelse av høyere kumulative doser av antiblast.
05.2 Farmakokinetiske egenskaper
Etter intravenøs infusjon av glutation i en dose på 2 g / m2 hos friske frivillige, økte den totale glutationkonsentrasjonen i plasma fra 17,5 ± 13,4 μmol / l (gjennomsnitt ± SD) til 823 ± 326 μmol / l. Distribusjonsvolumet for eksogent glutation ble beregnet til 176 ± 107 ml / kg og plasmahalveringstiden ble funnet å være 14,1 ± 9,2 minutter. Plasmacystein-konsentrasjonen økte fra 8,9 ± 3,5 mcmol / l 114 ± 45 mcmol / l etter infusjon. Til tross for økningen i cystein, reduserte den totale plasmakonsentrasjonen av totalt cystein, cystin og blandede disulfider, noe som indikerer en "økt passage av cystein inn i cellene."
Urinutskillelse av glutation og cyste (e) ina viste en økning på henholdsvis 300% og 10% i 90 minutter etter infusjonen.
Disse dataene indikerer at intravenøs administrering av glutation øker konsentrasjonen av sulfhydrylforbindelser i urinveiene markant og derfor også tilgjengeligheten av cystein på cellenivå. Den høye intracellulære konsentrasjonen av cystein rettferdiggjør den beskyttende effekten mot xenobiotika, ettersom den direkte eller indirekte oversettes til en økning i glutationbiosyntese.
05.3 Prekliniske sikkerhetsdata
Ikke-kliniske data viser ingen risiko for mennesker basert på konvensjonelle studier av sikkerhetsfarmakologi, toksisitet ved gjentatt dosering, gentoksisitet, reproduksjonstoksisitet.
06.0 LEGEMIDDELOPPLYSNINGER
06.1 Hjelpestoffer
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning"
Hetteglassene med pulver inneholder bare den aktive ingrediensen og løsningsmiddelampullene inneholder vann for injeksjonsvæsker.
TAD "2500 mg / 25 ml pulver og væske til infusjonsvæske, oppløsning"
Hetteglasset med pulver inneholder bare den aktive ingrediensen, og hetteglasset med løsningsmiddel inneholder vann for injeksjonsvæsker.
06.2 Uforlikelighet
I mangel av inkompatibilitetsstudier må legemidlet ikke blandes med andre produkter.
06.3 Gyldighetsperiode
3 år med henvisning til produktet i intakt emballasje, riktig lagret.
Den rekonstituerte løsningen er stabil i 8 timer ved romtemperatur.
06.4 Spesielle forholdsregler for lagring
Dette legemidlet krever ingen spesielle oppbevaringsbetingelser.
06.5 Emballasje og innhold i pakningen
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning" 5 hetteglass pulver + 5 hetteglass med løsningsmiddel 4 ml
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning" 10 hetteglass pulver + 10 hetteglass med løsningsmiddel 4 ml
Den aktive ingrediensen, i form av et hvitt lyofilisert pulver, finnes i hetteglass med lukkede og forseglede type III -glass.
Hetteglass med oppløsningsmiddel av glass inneholder vann til injeksjonsvæsker.
TAD "2500 mg / 25 ml pulver og væske til infusjonsvæske, oppløsning" 1 hetteglasspulver + 1 hetteglass med løsningsmiddel
Virkestoffet er inneholdt i et hetteglass av type III med en munndiameter på 29 mm, lukket og lukket.
Hetteglasset med løsningsmiddel, type I -glass, lokket og forseglet, inneholder vann for injeksjonsvæsker.
Pakken inneholder også et intravenøst infusjonssett som inneholder en tosidig pigg (overføringsenhet) for ekstern tilberedning av løsningen.
Det er ikke sikkert at alle pakningsstørrelser blir markedsført
06.6 Bruksanvisning og håndtering
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning"
Rekonstituer oppløsningen i hetteglasset med pulver ved å trekke ut vannet i hetteglasset med løsningsmiddel ved hjelp av en sprøyte utstyrt med en passende nål. Fjern aluminiumsfliken på hetteglasset og desinfiser proppen med en bomullspinne fuktet i alkohol, og sett deretter sprøytenålen inn inn i hetteglasset gjennom midten av gummiproppen og led vannstrømmen til glassveggen i hetteglasset. Rist forsiktig for å lette fullstendig oppløsning, og administrer deretter oppløsningen som er oppnådd langsomt intramuskulært eller intravenøst.
TAD "2500 mg / 25 ml pulver og væske til infusjonsvæske, oppløsning"
Fortsett først med rekonstituering av løsningen under aseptiske forhold ved å bruke følgende metoder:
1. Trekk ut aluminiumsfliken på hetteglasset med pulver og desinfiser hetten med en bomullspinne dyppet i alkohol
2. Fjern en enkelt hette fra engangs dobbeltspiss (hell) og sett spissen inn i hetteglasset med pulver gjennom midten av gummiproppen.
3. Trekk ut aluminiumsflippen på hetteglasset og desinfiser gummiproppen, fjern den andre hetten fra overføringsenheten og sett spissen inn i hetteglasset og snu den opp ned
4. Rist kort for å lette utstrømningen av vannet til hetteglasset med pulver, når hetteglasset er tømt, fjern skjenket og rist for å lette oppløsningen
Administrering av den rekonstituerte løsningen intravenøst utføres deretter som følger:
5.Desinfiser hetteglassproppen igjen, fjern deretter topplokket på slutten av infusjonssettet og sett det inn i midten av hetteglasset.
6. Påfør slangeklemmen og stram den helt på slangen;
7. Fjern nåleholderlokket og påfør en nål;
8. Trykk på dryppskålen for å fylle den omtrent halvveis, og åpne deretter slangeklemmen til all luft slipper ut fra settet;
9. Lukk slangeklemmen helt, stikk nålen inn i venen og åpne slangeklemmen sakte til ønsket strømning er oppnådd.
Bruk umiddelbart etter at beholderne er åpnet. Den rekonstituerte oppløsningen må være klar og fri for synlige partikler. Den er for en uavbrutt administrering og eventuelle rester kan ikke brukes.
Ubrukte produkter og avfall fra denne medisinen må kastes i henhold til lokale lovkrav.
07.0 INNEHAVER AV MARKEDSFØRINGSTILLATELSE
Biomedica Foscama Group S.p.A.
Via degli Office of the Vicar, 49
00186 Roma - (Italia)
08.0 NUMMER FOR MARKEDSFØRINGSTILLATELSE
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning" 5 hetteglass pulver + 5 hetteglass med løsningsmiddel 4 ml - AIC n. 027154032
TAD "600 mg / 4 ml pulver og væske til injeksjonsvæske, oppløsning" 10 hetteglass pulver + 10 hetteglass med løsningsmiddel 4 ml - AIC n. 027154044
TAD "2500 mg / 25 ml pulver og væske til infusjonsvæske, oppløsning" 1 hetteglasspulver + 1 hetteglass med løsningsmiddel 25 ml - AIC n. 027154057
09.0 DATO FOR FØRSTE GODKJENNELSE ELLER FORNYELSE AV GODKJENNINGEN
Dato for første markedsføring: 2. november 1989
10.0 DATO FOR REVISJON AV TEKSTEN
28. juli 2014