Hva er Marfan syndrom?
Marfan syndrom beskriver en kompleks arvelig lidelse i bindevev, som først og fremst påvirker øynene, det kardiovaskulære systemet og muskuloskeletale systemet. Med tanke på at hvert organ består av bindevev, kan Marfan syndrom ideelt ødelegge og forstyrre veksten og funksjonen til ethvert anatomisk sted.
Syndromet overføres som et autosomalt dominerende trekk: vi står derfor overfor en alvorlig genetisk sykdom som har et ekstremt variabelt "fenotypisk uttrykk (feilene kan variere enormt fra familie til familie eller fra pasient til pasient).
Det som utløser Marfan syndrom er endringen av FBN1-genet (på kromosom 15), som koder for fibrillin-1, et veldig viktig bindeglykoprotein som utgjør den strukturelle støtten til mikrofibriller.
Mikrofibriller: som består av fibrillin, er mikrofibriller tilstede i den ekstracellulære matrisen, der de danner en sammenfletting for avsetning av elastin i de elastiske fibrene. Selv om de er allestedsnærværende i kroppen, florerer mikrofibriller fremfor alt i aorta, leddbånd og sonuler i ciliary organer (på okulært nivå).
Siden dette er en autosomal dominerende sykdom, er det bare barn som har arvet et endret FBN-1-gen fra begge foreldrene som er påvirket av Marfan syndrom. Likevel er sykdommen i ett av fire tilfeller et resultat av spontane mutasjoner hos pasienter som ikke har en familiehistorie.
Navnet på sykdommen stammer fra den franske barnelegen som først beskrev det i 1896 (A. Marfan), hvoretter det var nødvendig å vente til 1991 for å identifisere det endrede genet som er involvert i den symptomatologiske manifestasjonen: oppdageren var F. Ramirez.
Se videoen
- Se videoen på youtube
Årsaker
Vi har nevnt at Marfans syndrom er det umiddelbare uttrykket for mutasjonen av et gen som koder for fibrillin-1.
FIBRILLIN 1 er en glykoproteinkomponent i elastin, avgjørende for å sikre og opprettholde vevets elastisitet og styrke. Under fysiologiske forhold binder fibrillin 1 seg til et annet protein, kjent som TGF-beta (eller transformerende vekstfaktor beta). TGF-beta ser ut til å være involvert i skadelige prosesser som påvirker vaskulær glatt muskel og den ekstracellulære matrisen. Ut fra disse antagelsene er noen forfattere overbevist om at Marfan syndrom, i tillegg til mutasjonen av FBN-1-genet, også skyldes et overskudd av TGF-beta, spesielt i aorta, hjerteventiler og lunger.

Forekomst
Det anslås at Marfan syndrom rammer 1 av hver 3.000-5.000 fødsler og forekommer vilkårlig mellom menn og kvinner. Statistikk viser at 75% av pasientene har en positiv familiehistorie; i de resterende 25% ligger årsaken til sporadiske mutasjoner som på en eller annen måte ser ut til å være forbundet med farens høye alder ved befruktningstidspunktet.
Barn med ekstremt alvorlige former for Marfan syndrom har en "forventet levetid på mindre enn ett år."
Før utviklingen av åpne hjertekirurgiske strategier hadde de fleste pasientene med Marfan syndrom en gjennomsnittlig levetid på 32 år; takket være den konstante forbedringen av medisinske og farmakologiske behandlinger, lever for tiden Marfan syndrom i gjennomsnitt opptil 60 år.
Tegn og symptomer
For ytterligere informasjon: Symptomer på Marfan syndrom
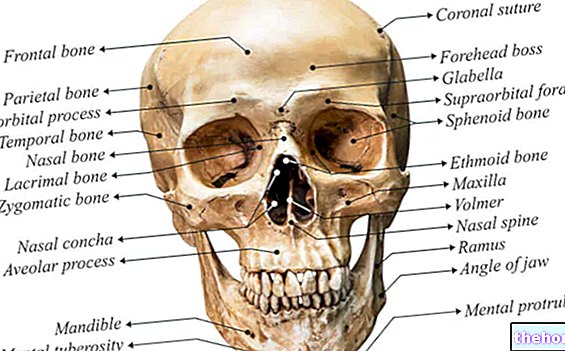
Marfan syndrom kan forekomme helt asymptomatisk. Berørte pasienter har en overdrevet slank struktur, som er uforholdsmessig høye og tynne. Nedre og øvre lemmer er mye lengre enn stammen (dolicostenomegali). Det er også snakk om arachnodactyly for best å uttrykke begrepet den overdrevne lengden på fingrene, typisk for de som er berørt av Marfan syndrom: hendene blir derfor sammenlignet med beina på en edderkopp.
Når det gjelder høyde, har disse pasientene en statur med et gjennomsnitt over 97. prosentilen.
Blant de andre særpregene som ofte er tilstede hos pasienter med Marfan syndrom, husker vi også:
- Åpning av armene større enn høyden
- Løse ledd → overdreven leddmobilitet
- Brystveggdeformitet
- Forskyvning av linsen
- Overkroppen er mindre utviklet enn det nedre området
- Spontan pneumothorax (11%)
- Skoliose
- Hudstriae på nivå med lår, rygg, deltoid, bryst
Blant de mest problematiske tegnene knyttet til Marfan syndrom, husker vi prolaps av hjerteventilen og mitralventilens mangel: en lignende tilstand kan lett favorisere utvidelsen av aortaringen og aortadisseksjonen.
Tabellen viser tegnene som kan finnes hos pasienter med Marfan syndrom. Karakterene beskrevet der er ikke alltid tilstede, men en god del av dem kan bli funnet.
Mulige symptomer
Hud
Striae i bryst-, kors- og sakralområdet
Øyne
Synsendring, astigmatisme, netthinneløsning, lukket vinkelglaukom, linselukasjon, nærsynthet
Bein struktur
Artralgi, kyfoskoliose, dolichostenomelia (overdreven utvikling i lengden på lemmer i forhold til bagasjerommet), hypermobilitet, høy gane, deformert bryst, flate føtter, stramme og tynne håndledd, unormal re-entry / protusion av brystbenet, skoliose, buede skuldre, spondylolistese
Fingre
arachnodactyly
Lunger
Spontan pneumothorax, dyspné, idiopatisk obstruktiv lungesykdom
Ansiktsendringer
Ogival gane (misdannelse av ganen), mandibular retrognathia (utviklingsdefekt i kjeven), langstrakt ansikt
Hjerte
Angina pectoris, abdominal aortaaneurisme, hjertearytmi, thorax aorta -dilatasjon / ruptur / disseksjon, aortainsuffisiens, mitralventilprolaps
Språk
Vanskeligheter med tale
Diagnose
Med tanke på de mer enn 200 mulige mutasjonene, er bruk av genetiske markører nesten umulig for diagnostiske formål.
Vurderingen av Marfans syndrom er ikke alltid så umiddelbar, siden fenotypisk uttrykk for mutasjonen ikke alltid er tydelig og lett å identifisere. Den diagnostiske forsinkelsen kan forringe pasientens overlevelse alvorlig: tenk for eksempel på manglende anerkjennelse av et kardiovaskulært problem.
De diagnostiske kriteriene for Marfan syndrom ble utarbeidet internasjonalt i 1996: diagnosen består av "undersøkelse av familiehistorien forbundet med en kombinasjon av store og mindre indikatorer av syndromet.
Noen av de mange diagnostiske testene som brukes er:
- ekkokardiogram
- magnetisk angioresonans og CT (for undersøkelse av aorta)
- magnetisk resonansangiografi (MRA) med kontrastvæske (for å markere de indre strukturene i aorta)
- undersøkelse med spaltelamper (for å analysere mulig forskyvning av linsen)
- måling av okulært trykk (for å markere den mulige tilstedeværelsen av glaukom)
- genetiske tester (anbefalt før et barn blir født for å finne ut om syndromet er eller ikke)
Terapier
Siden dette er en genetisk sykdom, er det ingen medisiner eller behandling som kan reversere sykdommen.
Bruk av medisiner er imidlertid avgjørende for å lindre symptomene og unngå komplikasjoner, spesielt hjertekomplikasjoner.For dette formålet er medisiner for å senke blodtrykket, for eksempel sartaner (fremfor alt), ACE -hemmere og betablokkere, spesielt egnet.
I forbindelse med Marfan syndrom kan pasienter som også lider av skoliose følge spesifikk behandling, så vel som for de som er rammet av DrDeramus.
Kirurgi kan tenkes for å korrigere den unormale aorta -dilatasjonen, et element som ofte forener de fleste pasienter med Marfan syndrom.
Fortsett: Marfan syndrom - medisiner og behandling "