En arvelig tilstand, SMA, skyldes mutasjoner i SMN1 -genet eller SMN2 -genet, hvis formål er å produsere et protein som tjener til å sikre overlevelse av motoriske nevroner.
Det er fem forskjellige former for spinal muskelatrofi: type 0, type 1, type 2, type 3 og type 4. De tre første typene er svært alvorlige og får pasienten til å dø for tidlig; type 3 og type 4 er mildere varianter, som påvirker pasientens levestandard, men uten å forårsake for tidlig død.
En genetisk test på en blodprøve er nødvendig for å diagnostisere SMA.
For tiden er behandlingen av SMA hovedsakelig basert på symptomatiske behandlinger, rettet mot å lindre lidelser og kontrollere komplikasjoner. En kur er tilgjengelig, basert på prinsippene for genterapi, men det er en veldig kostbar løsning og gjelder bare for visse pasienter.
, som manifesterer seg med atrofi og påfølgende svekkelse av skjelettmuskulaturen, og motoriske vansker.
SMA er en tilstand som kan forårsake pasientens død i ung eller veldig ung alder: de alvorligste sykdomsformene påvirker faktisk respirasjonsmusklernes effektivitet og er ansvarlige for episoder med respirasjonssvikt eller lungebetennelse dødelig utfall.
Motoriske nevroner og SMA

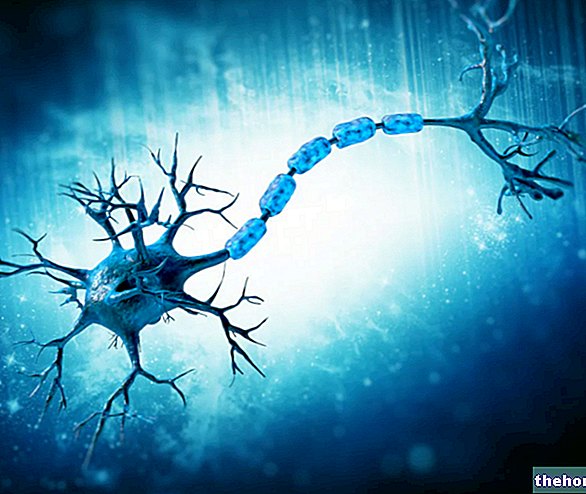
Motoriske nevroner, eller motoriske nevroner, er nerveceller som oppstår i sentralnervesystemet (hjerne og ryggmarg) og som ved hjelp av forlengelsene (aksonene) styrer aktiviteten til muskler og kjertler.
Det er to typer motoriske nevroner: øvre motoriske nevroner (eller første motoriske nevroner) og nedre motoriske nevroner (eller andre motoriske nevroner).
De øvre motoriske nevronene har sin opprinnelse i hjernen og styrer aktiviteten til de nedre motoriske nevronene, som hovedsakelig oppstår i ryggmargen og er ansvarlig for å lede aktiviteten til skjelettmuskulaturen (eller somatiske), i de glatte (eller viscerale) musklene, av hjertemuskelen og av hjertet.
Motorneuronene til personer med SMA degenererer gradvis og forårsaker "muskelatrofi fra inaktivitet, som i de alvorligste tilfellene resulterer i lammelse, respirasjonssvikt og død.
Epidemiologi: Hvor vanlig er spinal muskelatrofi?
SMA har en "årlig forekomst av 1 tilfelle per 10.000 nyfødte.
5 og som produksjonen av det såkalte overlevelsesproteinet til motoriske nevroner (SMN) avhenger av.Som navnet på proteinet produsert av SMN1 og SMN2 antyder, frata mutasjonen av disse genene motorneuronene et biologisk stoff som er avgjørende for deres overlevelse; mer presist, det reduserer proteinnivået: for eksempel i nærvær av mutasjoner i SMN1, faller nivåene av SMN-protein til 10-20% av det normale.
Det er klart at fraværet av tilstrekkelige mengder av SMN -proteinet bestemmer den progressive degenerasjonen av motoriske nevroner.
Tapet av motoriske nevroner avbryter nervesignalering som gjør det mulig å kontrollere aktiviteten til musklene i menneskekroppen; sistnevnte, som en konsekvens av det faktum at de ikke lenger er brukbare, gjennomgår en gradvis prosess med atrofi og svekkelse.
Visste du at ...
SMN2-genet er, for SMA, et sykdomsmodifiserende gen; Faktisk forekommer SMA i en mildere form hos pasienter med mutasjon i SMN1 og som av en eller annen grunn har tre eller fire kopier av SMN2 -genet.
Spinal muskelatrofi: Typer av mutasjon
Når SMA skyldes en "endring av SMN1, består den ansvarlige mutasjonen i 95-98% av tilfellene i en sletting av hele genet, mens det bare er i 2-5% i en" anomali av den normale gensekvensen.
Spinal muskelatrofi: en arvelig sykdom
I nesten alle tilfeller (98%) er den genetiske anomalien som er ansvarlig for SMA arvelig, det vil si at det er foreldrene til den syke personen som overfører den.
2% av ikke-arvelige tilfeller av SMA skyldes en mutasjon de novo skjedde på et veldig tidlig stadium av embryonal utvikling.
SMA og arvsmodell
Arvsmodellen for spinal muskelatrofi er autosomal recessiv, noe som betyr at for å arve SMA er det avgjørende at begge foreldre er friske bærere av den genetiske defekten i SMN1 eller SMN2, og at begge foreldrene gir den videre.
Når det gjelder autosomal recessive arvelige sykdommer som SMA, er sannsynligheten for at begge friske bærere vil overføre den genetiske defekten til barnet og dermed gjøre ham syk, 25%, eller én av hver fire tilfeller.
Typer SMA
Basert på begynnelsesalderen og alvorlighetsgraden av tilstanden, kjenner eksperter til fem forskjellige former for spinal muskelatrofi:
- SMA type 0: det er den mest alvorlige formen av alle. Det manifesterer seg allerede før fødselen med redusert mobilitet hos fosteret.
Spedbarn overlever vanligvis noen uker etter fødselen, selv når de får åndedrettsstøtte. - SMA type 1: av skjemaene som oppstår i løpet av livet, er det den mest alvorlige og vanligste (omtrent 50% av tilfellene); det vises i en tidlig alder, vanligvis innen den sjette måneden av livet.
Som regel er det dødsårsaken allerede i de første leveårene; sjelden, i ungdomsårene.
Døden oppstår vanligvis fra "respirasjonssvikt eller" lungeinfeksjon. - SMA type 2: det er formen som av tyngdekraften rangerer andre; Vanligvis begynner det mellom 7 og 18 måneders liv.
Forventet levetid for de som er rammet er større enn i det forrige tilfellet: Pasientene klarer faktisk å nå voksen alder. - SMA type 3: mindre alvorlig enn de to foregående, denne typen SMA oppstår vanligvis etter 18 måneders liv (i noen tilfeller kan den også vises i barndommen eller ungdomsårene).
Det innebærer store funksjonshemninger, men påvirker ikke levealderen. - SMA type 4: det er den voksne sykdomsformen, så vel som den minst alvorlige; det begynner vanligvis rundt det tredje tiåret av livet og har et veldig sakte forløp.
Det er generelt ikke ansvarlig for luftveisproblemer og er forbundet med en "normal levealder."
SMN -proteinnivåene påvirker alvorlighetsgraden av SMA: jo lavere mengde SMN, desto større er alvorlighetsgraden av den relaterte sykdommen.
Reduksjonen i SMN -nivåer er nært knyttet til omfanget av den genetiske defekten som påvirket SMN1- eller SMN2 -genene: jo mer omfattende denne defekten er, jo mer signifikant er reduksjonen i mengden av SMN -proteinet (dette er tilfellet for eksempel på en gen -sletting).
Videre kompromitterer ikke SMA de intellektuelle funksjonene (IQ hos pasienter er normal) og sparer syneorganet.
For ytterligere informasjon: SMA: alle symptomeneType 0 SMA symptomer
Som tidligere nevnt, forekommer type 0 SMA allerede i prenatal alder med redusert fostermobilitet; ved fødselen, viser det syke barnet tydelig problemer med å svelge og puste.
Sykdommen resulterer i død innen få uker etter fødselen, selv når pasienten får respiratorisk støtte.
Type 1 SMA symptomer
Barn med SMA type 1 har svært svake muskler som ikke utvikler seg som de skal (muskelsvinn). Dette forhindrer dem i å gjøre aktiviteter som å løfte hodet, bevege lemmene og innta en sittende stilling; dessuten kompliserer det gradvis viktige funksjoner, slik som å suge melk, svelge, tygge og puste.
Vanligvis er type 1 SMA dødelig i løpet av de første årene av livet; Noen pasienter klarer imidlertid å nå ungdomsalderen.
Døden oppstår vanligvis på grunn av respirasjonssvikt eller en "lungeinfeksjon på grunn av svelgevansker (inntak av lungebetennelse eller lungebetennelse ab ingestis).
Type 2 SMA symptomer
SMA type 2 manifesterer seg klassisk med:
- Mykhet i musklene i armer og ben;
- Rystelser i fingre og hender;
- Vanskeligheter med å innta sittestilling uavhengig (pasienten klarer imidlertid å opprettholde den);
- Vanskeligheter med å stå og gå
- Deformitet og leddproblemer;
- Pustevansker og svelging av mat;
- Skoliose (vises vanligvis senere).
Selv i denne situasjonen er pustevansker og svelging av mat årsaken til for tidlig død, som vanligvis oppstår i begynnelsen av voksen alder.
Type 3 SMA symptomer
Type 3 SMA forårsaker problemer med holdning og balanse, håndrystelser og problemer med å stå opp fra sittende stilling, gå, gå i trapper og løpe.
I begynnelsen krever ikke plagene støtte for bevegelse; senere, med degenerering av et større antall motoriske nevroner, blir krykker, turgåere og rullestoler grunnleggende.
Selv om det kan skje, er det svært sjelden at pasienter med SMA type 3 lider av pusteproblemer og svelger mat.
I nærvær av denne formen for SMA er forventet levetid normal, men med alle de nevnte problemene.
Type 4 SMA symptomer
Med voksen debut er SMA type 4 vanligvis assosiert med:
- Svekkelse av muskeltonus i armer og ben;
- Vanskelighetsgrad
- Risting og plutselige rykninger i musklene.
I utgangspunktet er de nevnte klagene moderate; i alderdommen blir de mer konsekvente.
Som type 3 SMA er type 4 SMA ikke en sykdom som kan påvirke pasientens forventede levetid.
SMA: når skal du oppsøke lege?
Alle foreldre som vet at de er en sunn bærer av SMA, anbefales på det sterkeste å konsultere en barnelege med ekspertise på genetiske sykdommer og en genetiker.
Hvis du ikke har denne typen informasjon, er det godt å vurdere barnets motoriske utvikling og funksjonene som livet avhenger av (for eksempel pust) måned for måned.
Sikkert, manglende evne til å sitte eller innta sittestillingen, vanskeligheten med å mate, tilstedeværelsen av respirasjonsunderskudd og en tynn og mindre tonet muskulatur enn for jevnaldrende utgjør alarmklokker.
Når det gjelder den voksne formen for SMA, mistenkes den mer eller mindre plutselige starten på muskelsvakhet og vanskeligheter med å gå.
Spinal muskelatrofi: komplikasjoner
De alvorligste formene for SMA kan føre til komplikasjoner som:
- Kvelning av mat. Det skyldes redusert evne til å tygge og spise mat.
- Respirasjonssvikt. Det er en konsekvens av manglende evne til å kontrollere aktiviteten til respiratoriske muskler.
- Lungebetennelse ab ingestis (eller inhalasjonspneumoni). Det oppstår når fremmed materiale som bærer patogener, for eksempel mat, spytt eller nesesekret, kommer inn eller akkumuleres i lungene.
Lungebetennelse ab ingestis det er et resultat av svelgevansker. - Lammelse som resulterer i bruk av rullestoler. Det skjer når sykdommen uimotståelig har kompromittert pasientens bevegelsesevner.
- Underernæring. Det er en annen konsekvens av svelgeproblemet: pasienten sliter faktisk med å mate ordentlig.
Det bør bemerkes at noen ganger kan tester som elektromyografi eller muskelbiopsi brukes under diagnosen SMA.
SMA: Fysisk undersøkelse og anamnese
Den fysiske undersøkelsen hos en pasient som kan lide av SMA innebærer en grundig analyse av symptomene og søk etter noen typiske tegn på sykdommen, for eksempel:
- Svakhet og ømhet i musklene;
- Plutselige muskelsammentrekninger
- Redusert eller fraværende senereflekser.
Når det gjelder sykehistorien, fokuserer dette imidlertid hovedsakelig på pasientens familiehistorie for å fastslå om et annet familiemedlem (foreldre, søsken, besteforeldre) klager eller klager over lignende symptom. Åpenbart det faktum at SMA er en arvelig sykdom, overført fra foreldre.
Selv om de ikke tillater å stille en endelig diagnose, kan fysisk undersøkelse og sykehistorie gi svært nyttig informasjon, som leder undersøkelser mot å utføre en genetisk test.
Det er klart at hvis pasienten er et lite barn, vil foreldrene samhandle med legen under sykehistorien.
SMA og genetisk test

Den genetiske testen for påvisning av SMA innebærer søk og studier av mutasjoner i SMN1 / SMN2 -genene i en prøve av blodceller fra pasienten.
Tilstedeværelsen av genetiske endringer betyr åpenbart sykdom.
Analysen av de påviste mutasjonene er avgjørende for å fastslå hvilken type spinal muskelatrofi som er tilstede og alvorlighetsgraden av tilstanden.
For å kjenne resultatene av den nevnte genetiske testen, er det generelt nødvendig å vente fra 3 til 4 uker (de nøyaktige ventetidene varierer avhengig av det genetiske senteret som utfører testen).
SMA: er prenatal diagnose mulig?
Det er mulig å diagnostisere SMA i prenatal alder.
For å gjøre dette trenger du en genetisk test på en prøve av fosterceller, innhentet ved delikate metoder som villocentese eller fostervannsprøve.
Gitt risikoen for abort som kjennetegner CVS og fostervannsprøve, driver legene med prenatal forskning for mutasjoner som kan tilskrives "spinal muskelatrofi bare hvis det er en familiehistorie med SMA bak det eller hvis det ufødte barnet er barnet til friske bærere av sykdommen.
SMA og nyfødt screening
Det skal bemerkes at i et par italienske regioner (Lazio og Toscana) er en tjeneste aktiv screening for tidlig diagnose av SMA og andre alvorlige genetiske sykdommer.
Den tidlige diagnosen av disse sykdommene tillater rettidig planlegging av den mest hensiktsmessige symptomatiske behandlingen for å kontrollere symptomer og komplikasjoner.
Spinal muskelatrofi og planlegging av graviditet
Genetisk rådgivning anbefales for alle kvinner som søker graviditet som:
- De hadde et barn med SMA i et tidligere svangerskap;
- De har en familiehistorie med SMA bak seg;
- Er de friske bærere av sykdommen eller er partneren deres.
Genetisk rådgivning kan hjelpe kvinner med disse forholdene til å forstå hvilke farer et fremtidig barn utsettes for.
SMA og differensialdiagnose
Det er to patologier som ligner veldig på SMA, som bare en "grundig diagnostisk undersøkelse gjenkjenner og forhindrer forvirring med" spinal muskelatrofi: disse er "spinal muskelatrofi med respiratorisk lidelse (SMARD) og" bulbo-spinal muskelatrofi (BSMA). Eller Kennedys sykdom); den første skyldes en mutasjon av IGHMBP2 -genet som ligger på kromosom 11, mens den andre skyldes en mutasjon av kjønnskromosom X.
og legemidler) godkjente Zolgensma, genterapimetoden for behandling av spinal muskelatrofi.
Zolgensma består av en svært avansert teknikk for molekylærbiologi, som inkluderer bruk av en virusvektor som er i stand til å sette inn en normal kopi av SMN1 / SMN2-genet i DNA som er tilstede inne i motorneuronene til en pasient.
Administrering av den nevnte virusvektoren skjer ved intravenøs injeksjon.
Zolgensma viste seg å være effektiv. Imidlertid, som forventet, har den to hovedgrenser som forhindrer bruk av den:
- Det er veldig dyrt. Det er snakk om millioner av euro;
- Det gjelder bare SMA -pasienter under 2 år.
Spinal muskelatrofi: Symptomatiske behandlinger
Symptomatiske terapier for SMA garanterer større fordeler hvis de tas i bruk umiddelbart; dette gjør tidlig diagnose av sykdommen svært viktig.
SMA og respiratorisk støtte
Riktig åndedrettsstøtte hjelper SMA -lider ikke bare med å puste, men også redusere risikoen for lungeinfeksjoner.
Blant de forskjellige terapeutiske alternativene er det masker for ikke-invasiv ventilasjon og mer invasive løsninger som orotrakeal intubasjon og trakeostomi; førstnevnte er ideelle for mindre alvorlige tilfeller, mens de mer invasive løsningene er avgjørende for pasienter med alvorlige problemer med luftveiene.
SMA og ernæringsstøtte
De alvorligste formene for spinal muskelatrofi påvirker evnen til å svelge og tygge mat, og utsetter pasienten for risiko for kvelning, lungebetennelse og underernæring.
For å kontrollere disse farlige konsekvensene er det avgjørende å ty til fôringshjelpemidler, for eksempel et nasogastrisk rør eller en gastrostomikirurgi, og stole på en ernæringsfysiolog som planlegger en diett som passer pasientens behov.
SMA og fysioterapi
De motoriske vanskene som kjennetegner pasienten med spinal muskelatrofi fører til ledd- og muskelstivhet fra inaktivitet.
Et tilstrekkelig fysioterapiprogram lar deg så langt som mulig forbedre musklernes fleksibilitet og gjøre leddene mindre stive.
Dette programmet inkluderer tydeligvis øvelser hvis utførelse er innenfor rekkevidden til pasientens evner.
SMA og ortopedi
I nærvær av skoliose, typisk for alvorlige former for SMA, er det viktig å konsultere en ortoped. sistnevnte kan indikere bruk av et ortopedisk korsett, hvis deformasjonen er mild, eller velge spinal fusjonskirurgi, hvis misdannelsen i ryggen er alvorlig.
Medisiner mot SMA
I noen år har det også vært spesifikke legemidler mot SMA.
Disse medisinene fortjener en egen behandling sammenlignet med symptomatiske terapier, selv om de ikke tillater å kurere sykdommen, men bare å inneholde den.
De spesifikke legemidlene mot SMA som er tilgjengelig for øyeblikket er Spinraza (nusinersen) og Evrysdi (risdiplam): de første handlingene ved å korrigere avvikende produksjon av SMN -proteinet i prosessen; den andre øker produksjonsnivået for SMN, og prøver også å holde dem på en kvote som er tilstrekkelig til behovene til den menneskelige organismen.
Godkjent av FDA i henholdsvis 2017 og 2020, garanterer Spinraza og Evrysdi resultater, i noen tilfeller enda mer enn tilfredsstillende, men de har en viktig begrensning: de er veldig dyre.
For mer informasjon: Spinraza: Hvordan det fungerer, risiko og fordeler
















-cos-sintomi-cause-e-gravidanza.jpg)