Generellitet
Retinoblastoma (Rb) er en ondartet øyetumor som utvikler seg fra cellene i netthinnen. Denne kreften kan oppstå i alle aldre, men utbruddet er mest vanlig i barndommen før fem år.

Barnekreft er aggressiv: Retinoblastom kan spre seg til lymfeknuter, bein eller benmarg. Sjelden involverer det sentralnervesystemet (hjerne og ryggmarg).
Omtrent 90% av barna med retinoblastom har en positiv prognose (sannsynlighet for helbredelse), forutsatt at diagnosen er tidlig og behandlingen startes før kreften sprer seg. Når det er mulig, er målet med medisinsk intervensjon å bevare pasientens syn.
Årsaker
Hendelsesrekken som fører til tumorutbrudd er kompleks. Dette begynner når celler i netthinnen utvikler en mutasjon (eller sletting), som involverer RB1 -tumorsuppressorgenet, plassert på q14 -båndet til kromosom 13 (13q14).
Hver celle har normalt to RB1 -gener:
- Hvis minst én kopi av genet fungerer som det skal, oppstår ikke retinoblastom (men risikoen øker);
- Når begge kopiene av genet er mutert eller mangler, oppstår ukontrollert celleproliferasjon.
I mange tilfeller er det uklart hva som egentlig induserer endringer i RB1 -genet (sporadisk retinoblastom); disse kan skyldes tilfeldige genetiske feil, som for eksempel oppstår under reproduksjon og celledeling. Imidlertid er det kjent at de genetiske abnormiteter som ligger til grunn for retinoblastom også kan overføres fra foreldre til barn, med et autosomalt dominerende arvemønster. Dette betyr at hvis en forelder bærer et mutert (dominerende) gen, vil hvert barn ha 50% sjanse for å arve det og 50% sjanse for å ha en normal genetisk sammensetning (recessive gener).
- En og annen celle inaktiverer den eneste normale kopien av RB1 -genet (en kopi er allerede mutert);
- Tapet av de to kopiene av RB1 fører til en "overdreven spredning av netthinnen.
- En og annen celle inaktiverer et av dets normale RB1 -gener;
- Den andre kopien av RB1 -genet er inaktivert;
- Tapet av de to kopiene av RB1 induserer en overdreven cellespredning som fører til retinoblastom.
Genetiske og molekylære egenskaper
- Retinoblastoma var den første svulsten som var direkte assosiert med en "genetisk abnormitet (sletting eller mutasjon av q14 -båndet til kromosom 13).
- RB1 koder for pRb -proteinet, som spiller en nøkkelrolle i cellesyklusen: det tillater DNA -replikasjon og cellesyklusprogresjon, ettersom det deltar i transkripsjonskontrollen av S -fasegener (G1 → † "S).
- I tillegg til retinoblastom, blir RB1 -genet inaktivert i blære-, bryst- og lungekreft.
Arvelig retinoblastom
Barn med arvelig retinoblastom har en tendens til å utvikle sykdommen i en tidligere alder enn sporadiske tilfeller. Videre har disse barna økt risiko for andre ikke-okulære kreftformer, ettersom abnormiteten i RB1-genet er medfødt (dvs. tilstede fra fødselen) og påvirker alle cellene i kroppen (kjent som en germline-mutasjon), inkludert de av begge. netthinne: Av denne grunn har barn med arvelig form ofte bilateralt retinoblastom i stedet for bare ett øye.
Symptomer
For å lære mer: Symptomer på Retinoblastoma
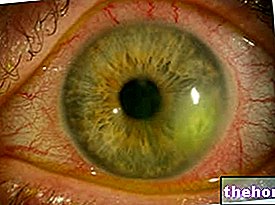
Det vanligste og mest åpenbare tegnet på retinoblastom er pupillens unormale utseende, som viser en gråhvit refleksjon når den blir rammet av en lysstråle (leukocoria eller amaurotisk kattrefleks). Andre tegn og symptomer inkluderer: nedsatt syn, øyesmerter og rødhet og forsinket utvikling. Noen barn med retinoblastom kan utvikle en myse (feiljusterte øyne); i andre tilfeller er det mulig å finne neovaskulær glaukom, som etter en stund kan forårsake forstørrelse av øyet (buftalmo).
Kreftceller kan ytterligere invadere øyet og andre strukturer:
- Intraokulært retinoblastom. Retinoblastom kan defineres som intraokulært når svulsten er helt inne i øyet. Neoplasma kan bare finnes i netthinnen eller også påvirke andre deler, for eksempel choroid, ciliary body og en del av synsnerven. Intraokulært retinoblastom spres derfor ikke til vevet rundt utsiden av øyet.
- Ekstraokulært retinoblastom.Svulsten kan formeres for å påvirke vevet rundt øyet (orbital retinoblastom). Kreften kan også spre seg til andre områder av kroppen, for eksempel hjernen, ryggraden, benmargen og lymfeknuter (metastatisk retinoblastom).
Tilstedeværelsen av orbital forlengelse, uveal involvering og invasjon av synsnerven er kjente risikofaktorer for utvikling av metastatisk retinoblastom.
Diagnose
Ved positiv familiehistorie gjennomgår pasienten regelmessige øyeundersøkelser for kreftscreening. Hvis medfødt retinoblastom er bilateralt, blir det vanligvis diagnostisert i det første leveåret, mens når det bare påvirker det ene øyet, kan tilstedeværelsen av svulsten bekreftes rundt 18-30 måneders alder.
Den kliniske diagnosen retinoblastom er etablert ved undersøkelse av fundus.Tumoren, avhengig av lokalisering, kan være synlig under en enkel undersøkelse av øyet, gjennom indirekte oftalmoskopi. Bildeteknikker kan brukes til å bekrefte diagnosen, definere iscenesettelsen av svulsten (hvor den er, hvor utbredt den er, om den påvirker funksjonene til andre organer i kroppen, etc.) og avgjøre om behandlingen har vært effektiv . Undersøkelser kan omfatte ultralyd, computertomografi (CT) og magnetisk resonansavbildning (MR).
Den molekylærgenetiske diagnosen er mulig gjennom identifisering av mutasjonen av RB1-genet. Den cytogenetiske analysen (dvs. av kromosomene) av perifere blodlymfocytter brukes til å oppdage sletting eller omorganisering som involverer kromosom 13 (13q14.1-q14. 2) .
Behandlinger
Ved retinoblastom kan flere behandlingsalternativer brukes.
Målet med behandlingen er:
- Eliminer svulsten og redde pasientens liv;
- Lagre øyet hvis mulig;
- Bevar syn så mye som mulig;
- Unngå utvikling av andre kreftformer, som også kan skyldes behandling, spesielt hos barn med arvelig retinoblastom.
Prognose (sannsynlighet for utvinning) og behandlingsalternativer avhenger av følgende faktorer:
- Stadium av svulsten;
- Pasientens alder og generelle helsetilstand;
- Plassering, størrelse og antall tumorfoci;
- Spredning av kreften til andre områder enn øyeeplet
- Hvor sannsynlig er det at synet kan bevares i det ene eller begge øynene.
De fleste tilfeller av retinoblastom diagnostiseres tidlig og behandles vellykket, før kreften kan metastasere utenfor øyeeplet, noe som resulterer i en kur på over 90%.